


2021-
Ana Da Lama, José Pérez Sestelo, Luis A. Sarandeses, and M. Montserrat Martínez
J. Org. Chem. 2024, doi.org/10.1021/acs.joc.3c02951
A new atom-economical synthesis of β-alkenyl-substituted BODIPYs via indium(III)-catalyzed intermolecular alkyne
hydroarylation with meso-substituted BODIPYs is described. While catalysis with InI3 allows the double β-functionalization of
BODIPY, resulting in regioselectively branched β,β′-disubstituted alkenyl BODIPYs, catalytic InCl3 enables the formation of linear
β-substituted alkenyl BODIPYs. Subsequent In(III)-catalyzed intermolecular alkyne hydroarylation allows the synthesis of
unsymmetrical push−pull BODIPY derivatives. Therefore, indium catalysis offers complementary regioselectivity in good chemical
yields and functional group tolerance. The resulting BODIPY dyes displayed bathochromically shifted absorption and emission
according to the electron-nature of the substituents in the alkenyl moiety with high molar extinction coefficients (ε up to 88,200 M−1
cm−1) and quantum yields (0.14−0.96).
Raquel Pérez-Guevara, Luis A. Sarandeses, Rosana Álvarez, M. Montserrat Martínez and José Pérez Sestelo
Adv. Synth. Cat. 2024, 366, 852 – 861 doi.org/10.1002/adsc.202301329
Tandem cycloisomerization reactions of functionalized 1,6-enynes under indium(III) catalysis are described. This atom-economic transformation proceeds smoothly with 5-exo-dig regioselectivity using commercial In(III) halides and 1,6-enynes furnished with alcohol, carboxylic acid or amine functional groups to give bicyclic structures in good yields and diastereoselectivities. The reaction with enynals involves a three–step mechanism to give an oxatricycle and a conjugated 1,3-diene. In the absence of the internal nucleophile the enyne cycloisomerization evolves through a skeletal rearrangement or a cyclopropanation reaction after the regioselective 5-exo-dig cyclization. The 1,6-enyne cycloisomerization is stereospecific and the stereoselectivity appears to be independent of the internal nucleophile. Experimental data support a common reaction mechanism involving an initial alkyne electrophilic p-coordination of In(III) followed by Markovnikov electrophilic alkene addition and ring-closure by nucleophilic attack. DFT studies hold up a stepwise mechanism involving the formation of a chiral non-classical carbocation intermediate that determines the diastereoselectivity of this tandem cycloisomerization reaction.
M. Montserrat Martínez, Jose Perez Sestelo, and Luis A. Sarandeses
Advances in Organometallic Chemistry 2023, 80, 177-253 doi.org/10.1016/bs.adomc.2023.02.001)
In this chapter the metal-catalyzed cross-coupling reactions of indium organometallics are reviewed. First, the various methods for the preparation of organoindium (transmetalation, direct insertion and indation of alkynes) are described and their structure are analyzed. Then, the reactivity in metal-catalyzed reactions is reviewed according to an organization based on the metal catalyst. The main section is the cross-coupling reaction under palladium catalysis which includes a general introduction, synthetic scope analysis, versatility and applications of the different indium organometallics and the reactive organic electrophiles (aryl, alkenyl, alkyl halides, triflates and others). Finally, other metal-catalyzed cross-coupling reactions are included along with general conclusions.
Overall, indium organometallics are shown as useful reagents for cross-coupling reactions with a variety of organic electrophiles. Their main features are the readily transmetalation to different transition metal complexes (palladium, nickel, rhodium, copper or iron) to transfer the organic groups attached to indium, and the high versatility and chemoselectivity as well.
Fabio Seoane-Carabel, Lorena Alonso-Marañón, Luis A. Sarandeses, José Pérez Sestelo
Synthesis 2023, 55, 1714-1723 (10.1055/s-0042-1751383)
1H-Isochromenes and 1,2-dihydroisoquinolines are synthesized
by regioselective indium(III)-catalyzed intramolecular hydrofunctionalization
of o-(alkynyl)benzyl derivatives. The reaction with o-
(alkynyl)benzyl alcohols and amines proceeds using indium triiodide
(5–10 mol%) in toluene at 80–100 °C via regioselective 6-endo-dig intramolecular
alkyne hydroalkoxylation or hydroamination in good yields.
Alternatively, the cycloisomerization reaction of o-(alkynyl)benzaldehydes
and imine derivatives using InI3 (5 mol%) and the Hantzsch ester
(120 mol%) takes place, under milder reaction conditions, to give a variety
of functionalized 1H-isochromenes and 1,2-dihydroisoquinolines
through a domino cycloisomerization/reduction approach.
Raquel Pérez-Guevara, Luis A. Sarandeses, M. Montserrat Martínez and José Pérez Sestelo
Org. Chem. Front., 2022, 9, 6894 (DOI: 10.1039/d2qo01600a)
The novel indium-catalyzed synthesis of benzannulated spiroketals by a double intramolecular hydroalkoxylation reaction of o-(hydroxyalkynyl)benzyl alcohols is reported. The reaction proceeds under mild reaction conditions using a low catalyst loading of indium triiodide (2–5 mol%) with a variety of benzyl
and homobenzyl alcohols, allowing the regioselective synthesis of a set of naturally occurring and new benzannulated spiroketals (5,5-, 5,6-, 5,7-, 6,6- and 6,7-) in good yields. The synthetic transformationinvolves the indium(III)-catalyzed electrophilic alkyne activation followed by the regioselective intramolecular hydroalkoxylation reaction. The method is the first example of spiroketal synthesis using main group catalysis representing an economical alternative to precious transition metals. In addition, it provides a complementary strategy for the regioselective synthesis of spiroketals and spiroaminals
Ana Da Lama, José Pérez Sestelo, Luis A. Sarandeses and M. Montserrat Martínez
Org. Biomol. Chem., 2022, 20, 9132–9137 DOI: https://doi.org/10.1039/d2ob01349e
A microwave-assisted one-pot synthesis of BODIPY dyes from pyrroles and acyl chlorides is reported. This protocol features short reaction times, low temperatures, minimum amount of solvent, scalability, versatility, and good yields of the products. These simple, efficient and sustainable conditions can be also
applied to the synthesis of derivatives such as BOPHY, BOAHY and BOPAHY.
Ana Da Lama, José Pérez Sestelo, Laura Valencia, David Esteban-Gómez, Luis
A. Sarandeses, M. Montserrat Martínez
Dyes and Pigments 205 (2022) 110539, DOI: 10.1016/j.dyepig.2022.110539
Fluorescent imidazole-triazole based ligands L1 and L2 have been designed as chemical push-pull chemosensors for divalent metal ions and synthesized through palladium-catalyzed cross-coupling reactions using indium organometallics and click chemistry. The novel ligands exhibit intense absorption in the ultraviolet region with high molar extinction coefficients, and strong fluorescence emission with large Stokes displacements.
On the basis of UV–Vis absorption spectroscopy and fluorescence emission data in acetonitrile, L1 is shown as a bifuncional chemosensor with differential response for Fe2+ and Cu2+ over a range of selected 3d divalent and other metal ions. The binding site of the ligand was established by single-crystal X-ray diffraction and 1H NMR
spectroscopy studies. The association constants, determined by spectrofluorimetric titrations, show a steady binding affinity of L1 for Cu2+ and Fe2+ in comparison with other previously reported fluorescent bidentate chemosensors, offering the lowest limit of detection (LOD) with Cu2+. DFT calculations provide a rationale properly understanding and interpreting the experimentally observed results. Indeed, a mechanism of the different optical responses of L1 towards 3d divalent metal ions is proposed.
Ana Da Lama, Beatrice Bartolomei, Cristian Rosso, Giacomo Filippini,
M. Montserrat Martínez, Luis A. Sarandeses, and Maurizio Prato
Eur. J. Org. Chem. 2022, e202200622, doi: 10.1002/ejoc.202200622
Boron-dipyrromethene complexes (BODIPYs) are attracting a growing interest for their notable photophysical properties.
Specifically, their application in organic photocatalysis is receiving particular attention thanks to their ability to undergo both electron and energy transfer processes, thus acting as promising photoredox catalysts and photosensitizers.
Although the number of examples employing these organic dyes is constantly increasing, there are not so many studies that correlate their chemical composition to their photocatalytic activity.
In this work, a rationally designed structure-activity relationship study was performed by selecting a synthetically relevant atom transfer radical addition (ATRA) reaction as a benchmark. We demonstrated how the presence of heavy atoms in the chromophore’s core turned out to be essential for achieving high reactivity levels. On the contrary, electronwithdrawing groups in position 8 (meso) undermined the catalytic performances, in agreement with the proposed reaction mechanism.
Chathurika R. K. Jayasundara, José M. Gil-Negrete, Jose R. Montero Bastidas, M. Montserrat Martínez, José Pérez Sestelo, Milton R. Smith, III, Robert E. Maleczka, Jr.
J. Org. Chem. 2022, 87, 751−759, DOI: 10.1021/acs.joc.1c01978
A versatile and efficient method to prepare borylated arenes furnished with alkyl, alkenyl, alkynyl, aryl and heteroaryl functional groups is developed by merging Ir-catalyzed C–H borylations (CHB) with a chemoselective palladium-catalyzed cross-coupling of triorganoindium reagents (Sarandeses-Sestelo coupling) with aryl halides bearing a boronic ester substituent. Using triorganoindium cross-coupling reactions to introduce unsaturated moieties enables the synthesis of borylated arenes that would be difficult to access through the direct application of the CHB methodology. The sequential double catalyzed procedure can be also performed in one vessel.
Ramón E. Millán, Jaime Rodríguez, Luis A. Sarandeses, Enrique Gómez-Bengoa, and José Pérez Sestelo
J. Org. Chem. 2021, 86, 10.1021/acs.joc.1c00825
The indium(III)-catalyzed cascade cycloisomerization reaction of 1,5-enynes with pendant aryl nucleophiles is reported. The reaction proceeds in cascade, under mild reaction conditions, using InI3 (5 mol%) as catalyst with a range of 1,5-enynes furnished with aryl groups (phenyl and phenol) at the alkene (E and Z isomers) and with terminal and internal alkynes. Using 1-bromo-1,5-enynes a one-pot sequential indium-catalyzed cycloisomerization and palladium-catalyzed cross-coupling with triorganoindium reagents was developed. The double cyclization is stereospecific and operates via a biomimetic cascade cation-olefin through a 1,5-enyne cyclization (6-endo–dig) and subsequent C–C hydroarylation or a C–O phenoxycyclization. DFT computational studies on 1,5-enynyl aryl ethers support a two-step mechanism where the first stereoselective 1,5-enyne cyclization produce a non-classical carbocation intermediate that evolves to the tricyclic reaction product through a SEAr mechanism. Using this approach, a variety of tricyclic heterocycles such as benzo[b]chromenes, phenanthridines, xanthenes, and spiroheterocyclic compounds are efficiently synthesized with high atom economy.
2016-2020
José Pérez Sestelo and Luis A. Sarandeses
Molecules 2020, 25, 4500; doi:10.3390/molecules25194500
Cross-coupling reactions stand among the most important reactions in chemistry [1,2]. Nowadays, they are a highly valuable synthetic tool used for the preparation of a wide variety of organic compounds, from natural and synthetic bioactive compounds to new organic materials, in all fields of chemistry [3]. Almost 50 years from its discovery, the research in this topic remains active, and important progresses are accomplished every year. For this reason, we believe that a Special Issue on this topic is of general interest for the chemistry community.
Gil-Negrete, J. M.; Pérez Sestelo, J.; Sarandeses, L. A.
J. Org. Chem. 2019, 84, 9778-9785. DOI: doi.org/10.1021/acs.joc.9b00928
Triorganoindium reagents (R3In) react with tetrahydroisoquinolines (THIQs) in the presence of Ph3CBF4 as oxidant to afford 1-substituted THIQs. The reaction proceeds in good yields at rt using 50 mol % of R3In with a variety of organic groups. 1H NMR studies show the generation of an iminium ion intermediate supporting a two-step mechanism based on THIQ oxidation and R3In nucleophilic addition. This reaction was applied to the synthesis of the alkaloid Nuciferine in three steps.

Pérez-Caaveiro, C.; Moreno Oliva, M.; López Navarrete, J. T.; Pérez Sestelo, J.; Martínez, M. M.; Sarandeses, L. A.
J. Org. Chem. 2019, 84, 8870-8885. DOI: http://doi.org/10.1021/acs.joc.9b00643
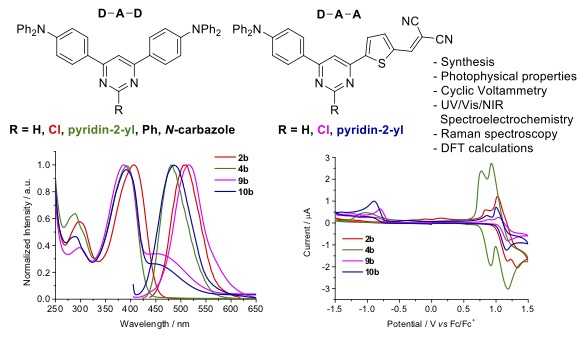
A series of donor-acceptor-acceptor (D-A-A) and donor-acceptor-donor (D-A-D) systems based on a pyrimidine -spacer with various substituents at C-2 position have been successfully prepared. The synthesis involved site-selective palladium cross-coupling reactions of chloropyrimidines with triorganoindium reagents and proceed in good yields and with atom economy. 4-(N,N-Diphenylamino)phenyl was chosen as donor group and thien-2-yl dicyanovinylene as acceptor one. The optical, vibrational, electrochemical and DFT calculations of these molecular systems were analyzed and experimental values show the important role of the substituents at C-2 position of the pyrimidine with stronger electron accepting ability, absorption in a wide range of UV-Vis, acceptable fluorescence lifetime and effective ICT properties. The ICT was observed in both series by the bathochromic shift on increasing the polarity of solvent. In addition, DFT calculations found a lower LUMOs of D-A-A molecules that suggest good electron ejection and transportation, being good properties for their application in various organic optoelectronic devices.
Pérez Sestelo, J. ; Sarandeses, L. A.; Martínez, M. M.; Alonso-Marañón, L.
Org. Biomol. Chem. 2018, 16, 5733–5747 (DOI: 10.1039/c8ob01426d).
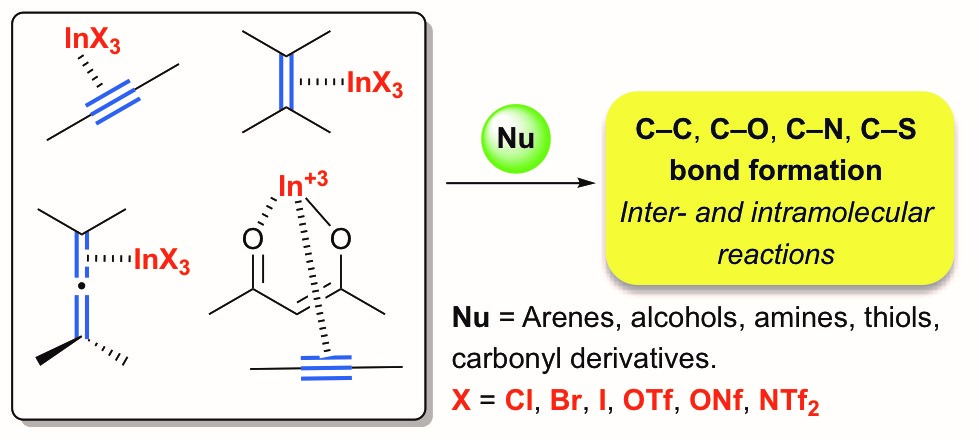
This review focuses on the utilization of indium(III) as p-acid for the activation of C–C unsaturated bonds in organic synthesis. In addition to its well-known s-coordination with carbonyl derivatives, indium(III) undergoes efficient p-coordination with unsaturated systems to trigger nucleophilic addition.
Accordingly, indium(III) halides and salts (InX3, X = Cl, Br, I, OTf, ONf, NTf2) have been reported as useful catalysts for a broad range of carbon–carbon and carbon–heteroatom bond formation reactions, including hydrofunctionalization (hydroarylation, hydroamination, hydroalkoxylation, and hydrothiolation), enyne cycloisomerization, and related reactions. In these reactions the counterion has a significant effect on the catalytic activity, and the development of novel In(III) complexes and the generation of highly electrophilic cationic indium(III) species has increased its synthetic applications as a p-acid catalyst.
Riveiros, R.; Tato, R.; Pérez Sestelo, J.; Sarandeses, L. A.
Molecules 2018, 23, 1582 (DOI: 10.3390/molecules23071582).

The activation of C–H bonds through catalytic reactions using transition metals is an important challenge in organic chemistry in which the intermediates are related to those produced in the classical cross-coupling reactions. As part of our research program devoted to the development of metal-catalyzed reactions using indium organometallics, a protocol for the C–H activation and C–C coupling of 2-arylpyridines with triorganoindium reagents under Rh(I) catalysis is reported. Under the optimized conditions, we found that Me3In and Ar3In reagents reacted with 2-arylpyridines and related compounds in the presence of Rh(PPh3)3Cl, in PhCl/THF (9:1), at 120 °C for 48 h, to afford the ortho -coupling products in moderate to good yields. The nitrogen atom in the pyridine ring acts as a directing group to assist the functionalization at the ortho position of the aryl group forming a new C–C bond at this position.
Alonso-Marañón, L.; Sarandeses, L. A.; Martínez, M. M.; Pérez Sestelo, J.
Org. Chem. Front. 2018, 5, 2308–2312 (DOI: 10.1039/c8qo00457a).
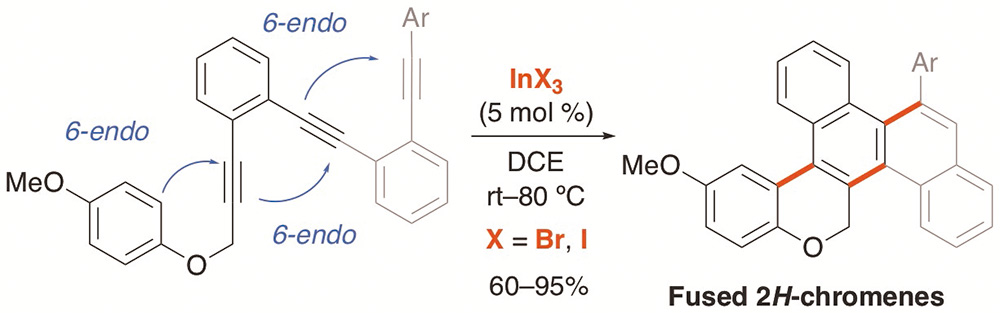
Fused 2H-chromenes are prepared by the cascade hydroarylation/cycloisomerization reactions of polyyne-type aryl propargyl ethers using indium(III) catalysis. The transformation proceeds with 6-endo-dig regioselectivity using InBr3 (5 mol%). The method can be extended to triynes allowing the formation of three bonds in one pot. Indium(III) also catalyzes the hydroamination/hydroarylation cascade reaction of o-aryldiynyl anilines to form fused carbazoles.
Alonso-Marañón, L.; Martínez, M. M.; Sarandeses, L. A.; Gómez-Bengoa, E.; Pérez Sestelo, J.
J. Org. Chem. 2018, 83, 7970–7980 (DOI: 10.1021/acs.joc.8b00829).
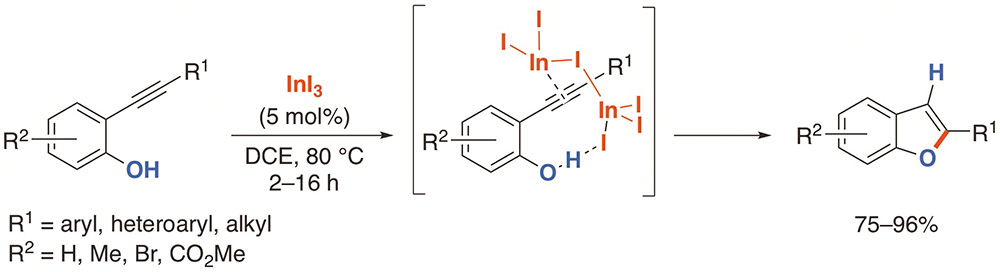
Indium(III) halides catalyze the hydroalkoxylation reaction of ortho-alkynylphenols to aff ord benzo[b]furans in good yields. The reaction proceeds with 5-endo-dig regioselectivity with a variety of phenols functionalized at the arene and alkyne moieties in high yields using InI3 (5 mol%) in DCE. Experimental and computational studies support a mechanism based on the indium(III) p-Lewis acid activation of the alkyne followed by nucleophilic addition of the phenol and final protodemetalation to afford the corresponding benzo[b]furan. DFT calculations suggest that dimer In2I6 is the catalytic species through a novel double coordination with the alkyne and the hydroxyl group.
Gil-Negrete, J. M.; Pérez Sestelo, J.; Sarandeses, L. A.
Chem. Commun. 2018, 54, 1453–1456 (DOI: 10.1039/c7cc09344f).

Highlighted in: Knochel, P.; Ketels, M. Synfacts 2018, 14, 414 (DOI: 10.1055/s-0037-1609133).
Bench-stable solid triorganoindium compounds have been prepared by coordination with 4-(dimethylamino)pyridine (DMAP). The solid R3In(DMAP) complexes are obtained from the corresponding solution of R3In in quantitative yield and can be stored for up to several weeks. These reagents show excellent reactivity in palladium-catalyzed cross-coupling reactions with organic electrophiles.
Mato, M.; Pérez-Caaveiro, C.; Sarandeses, L. A.; Pérez Sestelo, J.
Adv. Synth. Catal. 2017, 359, 1388–1393 (DOI: 10.1002/adsc.201601397).
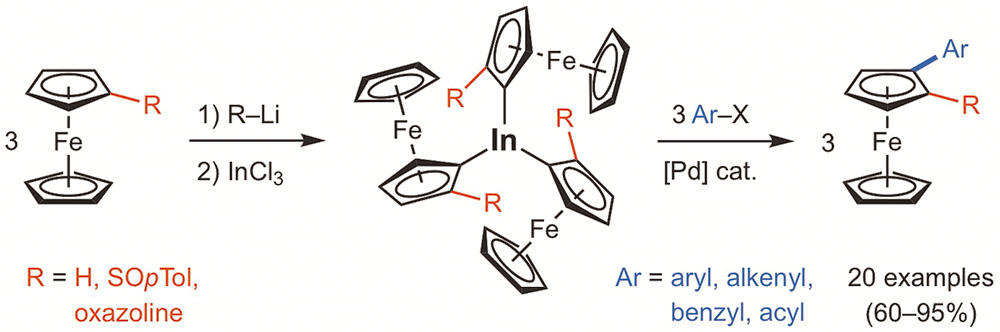
The preparation of ferrocenylindium species and palladium-catalyzed cross-coupling reactions for the synthesis of monosubstituted and planar chiral 1,2-disubstituted ferrocenes is described. Triferrocenylindium reagents (Fc3In) are efficiently prepared in a one-pot procedure from ferrocenes by lithiation and transmetallation to indium using InCl3 . The palladium-catalyzed cross-coupling reactions of Fc3In (40 mol%) with a variety of organic electrophiles (aryl, heteroaryl, benzyl, alkenyl and acyl halides) in THF at 80 °C overnight provided a wide variety of monosubstituted ferrocenes in good to excellent yields. This methodology allowed the stereoselective synthesis of planar chiral 2-aryl-1-oxazolylferrocenes and 2-aryl-1-sulfinylferrocenes, which are of interest in asymmetric catalysis.
Alonso-Marañón, L.; Sarandeses, L. A.; Martínez, M. M.; Pérez Sestelo, J.
Org. Chem. Front. 2017, 4, 500–505 (DOI: 10.1039/C6QO00721J).
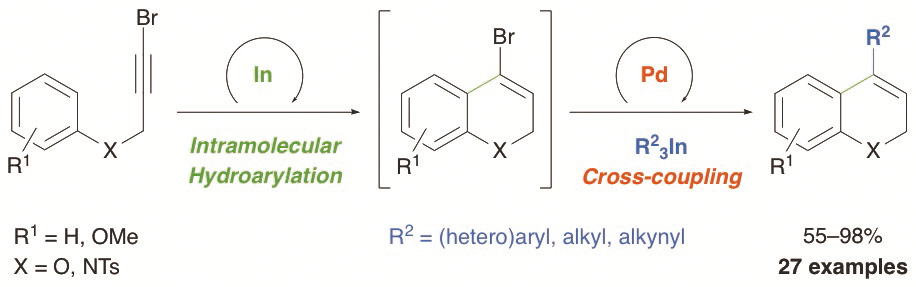
A sequential one-pot indium-catalyzed intramolecular hydroarylation (IMHA) of bromopropargyl aryl ethers and amines, and palladium-catalyzed cross-coupling reaction using triorganoindium reagents (R3In) has been developed. In this transformation, the IMHA of 3-bromo-2-propynyl aryl ethers under indium(III) catalysis, proceeds regioselectively through a 6-endo-dig pathway to afford 4-bromo-2H-chromenes. Subsequent palladium-catalyzed cross-coupling with R3In gives 4-substituted-2H-chromenes in one-pot. This sequential transformation was extended to 3-bromo-2-propynyl-N-tosylanilines to afford 4-substituted-1,2-dihydroquinolines. The dual-catalyzed procedure takes place efficiently with a variety of propargyl aryl ethers and amines and R3In (R = aryl, heteroaryl, alkyl or alkynyl), showing the efficiency of these organometallics and proving the compatibility of indium and palladium in catalysis.
Gil-Negrete, J. M.; Pérez Sestelo, J.; Sarandeses, L. A.
Org. Lett. 2016, 18, 4316–4319 (DOI: 10.1021/acs.orglett.6b02058).
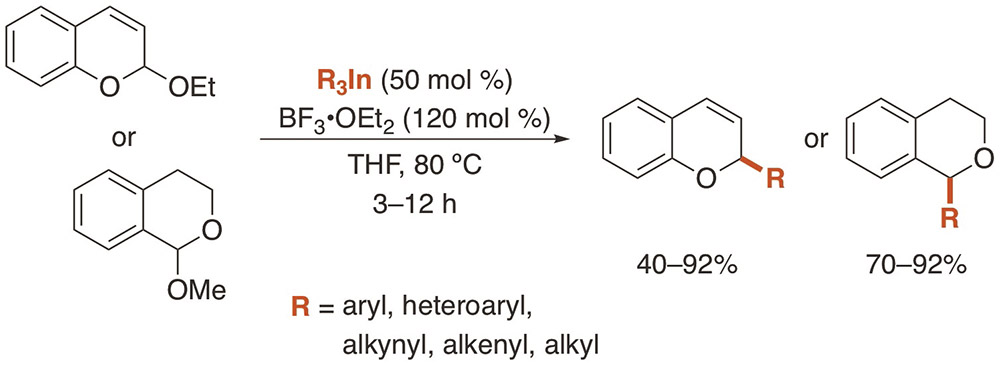
Highlighted in: Knochel, P.; Hammann, J. M. Synfacts 2016, 12, 1185 (DOI: 10.1055/s-0036-1589323).
A transition-metal-free coupling of triorganoindium reagents with benzopyranyl acetals mediated by a Lewis acid has been developed. The reaction of R3In with chromene and isochroman acetals in the presence of BF3·OEt2 afforded 2-substituted chromenes and 1-substituted isochromans, respectively, in good yields. The reactions proceed with a variety of triorganoindium reagents (aryl, heteroaryl, alkynyl, alkenyl, alkyl) using only 50 mol% of the organometallic, thus demonstrating the efficiency of these species. Preliminary mechanistic studies indicate the formation of an oxocarbenium ion intermediate in the presence of the Lewis acid.
2011-2015
Alonso-Marañón, L.; Martínez, M. M.; Sarandeses, L. A.; Pérez Sestelo, J.
Org. Biomol. Chem. 2015, 13, 379–387 (DOI: 10.1039/c4ob02033b).
Highlighted in: Snieckus, V.; Maheta, A. Synfacts 2015, 11, 255 (DOI: 10.1055/s-0034-1380240).

Indium(III) halides catalyze efficiently the intramolecular hydroarylation (IMHA) of aryl propargyl ethers. The reaction proceeds regioselectively with terminal and internal alkynes bearing electron-rich and electron-deficient substituents in the benzenes and alkynes affording only the 6-endo-dig cyclization product. Additionally, a sequential indium-catalyzed IMHA and palladium-catalyzed Sonogashira coupling can be performed in one reaction vessel. Experiments with deuterium support a mechanism through electrophilic aromatic substitution.
Mosquera, Á.; Férnandez, M. I.; Canle L., M.; Pérez Sestelo, J.; Sarandeses, L. A.
Chem. Eur. J. 2014, 20, 14524–14530 (DOI: 10.1002/chem.201403736).

The synthesis and photochemical study of novel nonsymmetrical 1,2-dithienylethenes (DTEs) with a maleimide bridge have been carried out. The synthetic approach to the DTEs was based on successive selective palladium-catalyzed cross-coupling reactions of 5-susbtituted-2-methyl-3-thiophenyl indium reagents with 3,4-dichloromaleimides. The required organoindium reagents were prepared from 2-methyl-3,5-dibromothiophene by a selective (C-5) coupling reaction with triorganoindium compounds (R3In) and subsequent metal–halogen exchange. The coupling reactions usually gave good yields and have a high atom economy with substoichiometric amounts of R3In. The results of photochemical studies show that these novel dithienylmaleimides undergo a photocyclization reaction upon irradiation in the UV region and a photocycloreversion after excitation in the visible region, thus they can be used as photochemical switches. ON–OFF operations can be repeated in successive cycles without appreciable loss of effectiveness in the process.
Pérez-Caaveiro, C.; Pérez Sestelo, J.; Martínez, M. M.; Sarandeses, L. A.
J. Org. Chem. 2014, 79, 9586–9593 (DOI: 10.1021/jo501664p).
Highlighted in: Knochel, P.; Hammann, J. M. Synfacts 2015, 11, 75 (DOI: 10.1055/s-0034-1379660).
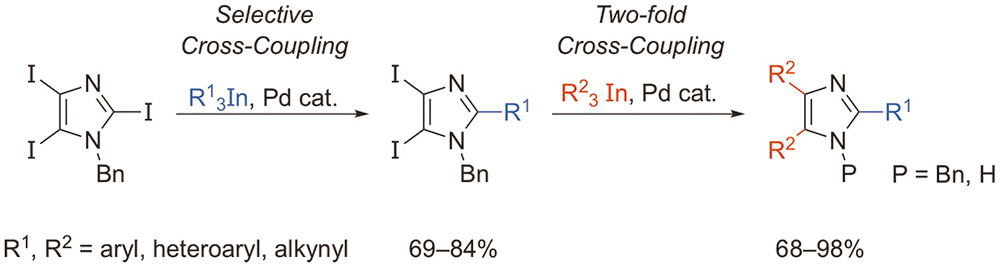
Triorganoindium reagents (R3In, R = aryl, heteroaryl, alkynyl) react selectively under palladium catalysis with N-benzyl-2,4,5-triiodoimidazole to afford the C-2 monocoupling products. The reaction proceeds efficiently for a variety of aryl- and heteroarylindium reagents with the transfer of all three organic groups attached to the metal. The coupling products can be used in a subsequent two-fold cross coupling to give trisubstituted imidazoles in good yields. This approach was employed to synthesize neurodazine and analogues in good yields.
Mosquera, A.; Pena, M. A.; Pérez Sestelo, J.; Sarandeses, L. A.
Eur. J. Org. Chem. 2013, 2555–2562 (DOI: 10.1002/ejoc.201300042).
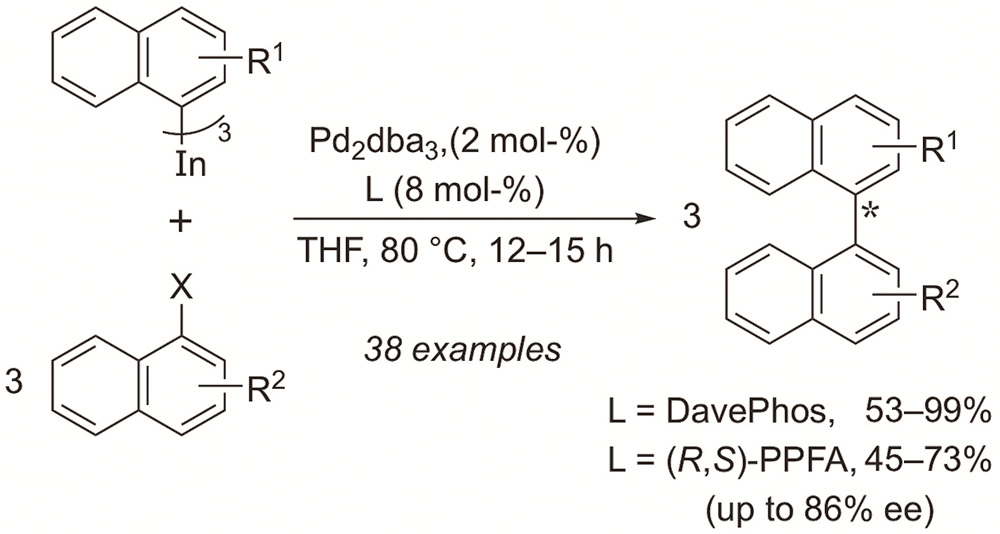
1,1’-Binaphthalenes and heterocyclic analogues can be efficiently prepared by palladium-catalysed cross-coupling reactions between tri(1-naphthyl)indium reagents and 1-halonaphthalenes and haloisoquinolines. The reactions were usually carried out in THF at 80 °C with a slight excess of the indium reagent (40 mol-%) and a low catalyst loading (4 mol% Pd) to afford the cross-coupling products in good yields (45–99%). The method allows the synthesis of sterically hindered 2-substituted and 2,2’-disubstituted 1,1’-binaphthalenes and naphthylisoquinolines. In addition, the coupling reactions can be performed enantioselectively and the best enantiomeric excesses were obtained by using the chiral amino-phosphane ferrocenyl ligand (R,S)-PPFA.
Peña-López, M.; Sarandeses, L. A.; Pérez Sestelo, J.
Eur. J. Org. Chem. 2013, 2545–2554 (DOI: 10.1002/ejoc.201201720).

Cross-coupling reaction of organogold(I) phosphanes with organic electrophiles in aqueous media has been investigated. Reactions between isolated aryl-, alkenyl-, or alkynylgold(I) phosphanes and aryl halides or triflates, alkenyl halides, and allyl acetates proceed under palladium catalysis conditions at room temperature or 80 °C in water with THF as a co-solvent. The coupling reactions give good yields and are highly versatile and chemoselective, allowing the presence of free amino or hydroxy groups in the electrophile. This methodology was applied to the preparation of substituted phenylalanine esters in a demonstration that gold(I) organometallics are suitable reagents for metal-catalyzed cross-coupling reactions under protic conditions.
Martínez, M. M.; Pérez-Caaveiro, C.; Peña-López, M.; Sarandeses, L. A.; Pérez Sestelo, J.
Org. Biomol. Chem. 2012, 10, 9045–9051 (DOI: 10.1039/c2ob26398j).

4,6-Disubstituted-2-(4-morpholinyl)pyrimidines, an important class of bioactive compounds, have been synthesized from 4,6-dichloro-2-(4-morpholinyl)pyrimidine by selective and sequential palladium catalyzed cross-coupling reactions using triorganoindium reagents. This methodology, being efficient and versatile, allowed the synthesis of a variety of non-symmetrical pyrimidines functionalized at C-4 and C-6 positions.
Riveiros, R.; Tato, R.; Pérez Sestelo, J.; Sarandeses, L. A.
Eur. J. Org. Chem. 2012, 3018–3023 (DOI: 10.1002/ejoc.201200104).
A novel rhodium-catalyzed allylic substitution reaction using indium organometallics is reported. Aryl- and heteroarylindium reagents reacted in THF at 80 °C with primary and secondary allyl halides and their derivatives under rhodium(I) catalysis to afford the α-substituted products in good yields and with high regio- and stereoselectivity. The reaction takes place with substoichiometric amounts of the triorganoindium reagents, which demonstrates the efficiency of the indium–rhodium transmetallation process in carbon–carbon bond-forming reactions.
Martínez, M. M.; Peña-López, M.; Pérez Sestelo, J.; Sarandeses, L. A.
Org. Biomol. Chem. 2012, 10, 3892–3898 (DOI: 10.1039/c2ob25123j).
The synthesis of unsymmetrical 2,5-disubstituted thiophenes by selective and sequential palladiumcatalyzed cross-coupling reactions of indium organometallics with 2,5-dibromothiophene is reported. Following an iterative coupling sequence, a-oligothiophenes were synthesized in good yields and with high atom economy.
Peña-López, M.; Ayán-Varela, M.; Sarandeses, L. A.; Pérez Sestelo, J.
Org. Biomol. Chem. 2012, 10, 1686–1694 (DOI: 10.1039/c2ob06788a).
Aryl and alkenylgold(I) phosphanes react regioselectively with allylic electrophiles such as cinnamyl and geranyl halides (bromide, chloride and acetates) under palladium catalysis in THF at 80 °C to afford the α-substitution product with moderate to high yields. When the reaction is performed with a chiral enantiopure secondary acetate, the α-substituted cross-coupling product is obtained with complete inversion of the stereochemistry.
Tato, R.; Riveiros, R.; Pérez Sestelo, J.; Sarandeses, L. A.
Tetrahedron 2012, 68, 1606–1611 (DOI: 10.1016/j.tet.2011.11.075).
A novel rhodium-catalyzed conjugate addition of indium reagents to electron deficient olefins is reported. The reaction takes place in THF/MeOH at 110 °C using arylindium dichlorides, a rhodium(I)-binap complex as catalyst, and α,β-unsaturated ketones and lactones in good yields (45–94%). The addition of MeOH is crucial for an efficient transformation and NMR studies seem to indicate that promotes the catalytic cycle leaving the indium organometallic unaltered. The use of chiral non-racemic ligands allows performing the reaction with moderate enantioselectivities.
2006-2010
Peña-López, M.; Ayán-Varela, M.; Sarandeses, L. A.; Pérez Sestelo, J.
Chem. Eur. J. 2010, 16, 9905–9909 (DOI: 10.1002/chem.201000726).
The palladium-catalyzed cross-coupling reaction of organogold(I) reagents (alkyl, alkenyl, aryl, and alkynyl) with organic electrophiles, such as aryl and alkenyl halides, aryl triflates, benzyl bromide, and benzoyl chloride is reported. The reaction takes place, under palladium catalysis, at room temperature with short reaction times to give the corresponding cross-coupling products in high yields.
Peña-López, M.; Martínez, M. M.; Sarandeses, L. A.; Pérez Sestelo, J.
J. Org. Chem. 2010, 75, 5337–5339 (DOI: 10.1021/jo100779z).
Fumaquinone, a novel prenylated naphthoquinone antibiotic, was synthetized from ethyl acetoacetate in three steps (58% overall yield). The key step of the synthesis is the construction of the naphthoquinone skeleton by a regioselective Diels- Alder reaction between a 2-alkyl 1,3-bis(trimethylsilyloxy)-1,3-diene derivative and a bromoquinone. This short and versatile approach confirms the structure of fumaquinone and allows the synthesis of derivatives at the C-6 position.
Peña-López, M.; Martínez, M. M.; Sarandeses, L. A.; Pérez Sestelo, J.
Org. Lett. 2010, 12, 852–854 (DOI: 10.1021/ol902920u).
Addition/Correction: Org. Lett. 2011, 13, 4151–4151 (DOI: 10.1021/ol201592g).
A versatile enantioselective total synthesis of barrenazines A and B has been accomplished from 1,4-butanediol. The key steps of the synthesis are a sequential allylboration/ring-closing metathesis for the construction of the tetrahydropyridine ring and the preparation of a functionalized 4-azidopiperidin-5-one through a stereoselective epoxidation and regioselective ring-opening reaction. The C2-symmetrical pyrazine skeleton of barrenazines was prepared by dimerization of the azidopiperidinone, and the carbon side chain was completed by copper-catalyzed reactions using Grignard reagents.
Bouissane, L.; Pérez Sestelo, J.; Sarandeses, L. A.
Org. Lett. 2009, 11, 1285–1288 (DOI: 10.1021/ol900063p).
Unsymmetrical 3,4-disubstituted maleimides have been synthesized by palladium-catalyzed cross-coupling reactions of indium organometallics with 3,4-dihalomaleimides. The synthesis was performed by stepwise or sequential one-pot palladium-catalyzed cross-coupling reactions with various triorganoindium reagents. This method was used to prepare a wide variety of alkyl, aryl, heteroaryl, and alkynyl 3,4-disubstituted maleimides in good yields and with high selectivity and atom economy.
Peña-López, M.; Martínez, M. M.; Sarandeses, L. A.; Pérez Sestelo, J.
Chem. Eur. J. 2009, 15, 910–916 (DOI: 10.1002/chem.200802021).
The first total synthesis of (+)-neomarinone has been achieved by following a concise and convergent route using methyl (R)-lactate and (R)-3-methylcyclohexanone as chiral building blocks. Key steps of the synthesis are the stereocontrolled formation of the two quaternary stereocenters by diastereoselective 1,4-conjugate addition and enolate alkylation reactions, and the construction of the furanonaphthoquinone skeleton by regioselective Diels–Alder reaction between a 1,3-bis(trimethylsilyloxy)-1,3-diene and a bromoquinone. The synthesis proves the relative and absolute stereochemistry of natural neomarinone.
Mosquera, A.; Riveiros, R.; Pérez Sestelo, J.; Sarandeses, L. A.
Org. Lett. 2008, 10, 3745–3748 (DOI: 10.1021/ol801393n).
The palladium-catalyzed cross-coupling reaction of triorganoindium reagents (R3In) with 5-bromo-2-chloropyrimidine proceeds chemoselectively, in good yields, to give 5-substituted-2-chloropyrimidines or 2,5-disubstituted pyrimidines using 40 or 100 mol% of R3In, respectively. Sequential cross-couplings are also performed, in one pot, using two different R3In. This method was used to achieve the first synthesis of the alkaloid hyrtinadine A. The key step was a two-fold cross-coupling reaction between a tri(3-indolyl)indium reagent and 5-bromo-2-chloropyrimidine.
Riveiros, R.; Saya, L.; Pérez Sestelo, J.; Sarandeses, L. A
Eur. J. Org. Chem. 2008, 1959–1966 (DOI: 10.1002/ejoc.200701216).
The regio- and stereoselectivity of the palladium-catalysed cross-coupling reactions of indium organometallics with stereodefined 1-haloalkenes and 1,1-dihaloalkenes have been studied. Triorganoindium reagents (R3In; R = alkyl, alkenyl, aryl and alkynyl) can be stereospecifically coupled with stereodefined alkenyl iodides in good yields and short reaction times under palladium catalysis. Additionally, the palladium- catalysed cross-coupling reaction of R3In (90 mol%) with 1,1-dibromo-1-alkenes gave dicoupling products in high yields. When the reaction was performed with 40 mol% of aryl-, vinyl- and alkynylindium derivatives, trans-selective monosubstitution products were obtained in moderate to good yields. These selective couplings were performed with [Pd2dba3]/P(2-furyl)3 (1:1, 2 mol%) at 0 °C or, for 1,1-dibromo-1-alkenes with an aromatic group in the b-position, Pd(DPEPhos)Cl2] (2 mol%) at room temperature as the catalytic system. The resulting (Z)-monobromoalkenes can be further functionalized by cross-coupling reaction with various R3In (R = alkyl, aryl and alkynyl) in the presence of [Pd(tBu3P)2] as catalyst, at room temperature, to provide trisubstituted olefins in good yields.
Caeiro, J.; Pérez Sestelo, J.; Sarandeses, L. A.
Chem. Eur. J. 2008, 14, 741–746 (DOI: 10.1002/chem.200701035).
Martínez, M. M.; Sarandeses, L. A.; Pérez Sestelo, J.
Tetrahedron Lett. 2007, 48, 8536–8538 (DOI: 10.1016/j.tetlet.2007.09.145).
A short enantioselective synthesis of barrenazines A and B is described. Barrenazines A and B are prepared following a common synthetic route in nine steps (19% overall yield) and eight steps (21% overall yield), respectively, from readily available 4-methoxy-3-(triisopropylsilyl)pyridine. The synthesis relies on a highly diastereoselective nucleophilic addition of a Grignard reagent to a chiral acylpyridinium salt, a radical azidation of a silyl enol ether and the assembly of the pyrazine ring by reductive dimerization of a functionalized 5-azidopiperidin-4-one.
Riveiros, R.; Pérez Sestelo, J.; Sarandeses, L. A.
Synthesis 2007, 3595–3598 (DOI: 10.1055/s-2007-983849).
Allenes have been efficiently prepared by the reaction of propargylic esters (benzoates, acetates, carbonates) with triorganoindium compounds (R3In) under palladium catalysis, via an SN2’ rearrangement. The reaction proceeds smoothly at room temperature with a variety of aryl-, alkenyl-, and alkynylindium reagents. The yields obtained are high and the regioselectivity is complete both in the case of terminal and nonterminal propargylic esters.
Suárez, R. M.; Martínez, M. M.; Sarandeses, L. A.; Pérez Sestelo, J.
Tetrahedron Lett. 2007, 48, 6493–6495 (DOI: 10.1016/j.tetlet.2007.07.064).
A practical and efficient stereoselective synthesis of the side chain of neomarinone is reported. The synthesis was achieved in six steps (41% overall yield) from 2-methyl-2-cyclohexenone. The key step is a novel stereoselective 1,4-conjugate addition/enolate alkylation by an epoxide-opening reaction.
Pena, M. A.; Pérez Sestelo, J.; Sarandeses, L. A.
J. Org. Chem. 2007, 72, 1271–1275 (DOI: 10.1021/jo062148s).
Highlighted in: Knochel, P.; Thaler, T. Synfacts 2007, 531 (DOI: 10.1055/s-2007-968435).
A range of biaryl compounds (aryl- aryl, aryl- heteroaryl, and heteroaryl- heteroaryl) can be efficiently prepared by a palladium-catalyzed cross-coupling reaction between ortho-substituted triarylindium reagents and aryl halides. The triarylindium reagents are prepared by directed ortho-lithiation and transmetallation to indium from the corresponding benzene derivatives using various directed metallation groups (DMGs). The reaction proceeds smoothly in high yields and short reaction times with high atom economy (the three aryl groups attached to indium are efficiently transferred).
Riveiros, R.; Rodríguez, D.; Pérez Sestelo, J.; Sarandeses, L. A.
Org. Lett. 2006, 8, 1403–1406 (DOI: 10.1021/ol060192o).
Triorganoindium reagents (R3In) react with propargylic esters under palladium catalysis via an SN2’ rearrangement to afford allenes in good yields and with high regioselectivity. The reaction proceeds smoothly at room temperature with a variety of R3In (aryl, alkenyl, alkynyl, and methyl). When chiral, nonracemic propargylic esters are employed, the reaction takes place with high anti-stereoselectivity providing allenes with high enantiomeric excess.
2001-2005
Pena, M. A.; Pérez Sestelo, J.; Sarandeses, L. A.
Synthesis 2005, 485–492 (DOI: 10.1055/s-2004-834945).
The use of indium organometallics in multifold and sequential cross-coupling reactions is reported. Triorganoindium reagents (R3In) react, under palladium catalysis, with oligohaloarenes affording the multiple cross-coupling products in a single operation. In the reaction, the three organic groups (alkyl, aryl, alkenyl or alkynyl) attached to indium are efficiently transferred to the electrophile, with only a slight excess of organometallic reagent. We demonstrate that indium organometallics are useful reagents for sequential cross-coupling reactions. This reaction illustrates the high chemoselectivity of R3In.
Suárez, R. M.; Pérez Sestelo, J.; Sarandeses, L. A.
Org. Biomol. Chem. 2004, 2, 3584–3587 (DOI: 10.1039/b413017k).
Enantiomerically pure natural and unnatural α-amino acids have been synthesized from a chiral methyleneoxazolidinone by means of a highly diastereoselective 1,4-conjugate addition of alkyl iodides in aqueous media. The zinc–copper conjugate addition reaction exhibits high chemoselectivity, with the possibility of using functionalized iodides, to afford a single diastereomer in short reaction times and with good yields.
Rodríguez, D.; Pérez Sestelo, J.; Sarandeses, L. A.
J. Org. Chem. 2004, 69, 8136–8139 (DOI: 10.1021/jo0491511).
The palladium(0)-catalyzed cross-coupling reaction of allylic halides and acetates with indium organometallics is reported. In this synthetic transformation, triorganoindium compounds and tetraorganoindates (aryl, alkenyl, and methyl) react with cinnamyl and geranyl halides and acetates to afford the SN2 product regioselectively and in good yield. The reaction proceeds with net inversion of the stereochemical configuration.
Cornella, I.; Suárez, R. M.; Mouriño, A.; Pérez Sestelo, J.; Sarandeses, L. A.
J. Steroid Biochem. Mol. Biol. 2004, 89-90, 19–23 (DOI: 10.1016/j.jsbmb.2004.03.045).
The synthesis of vitamin D3 active metabolites [24R,25-(OH)2-D3, 24S,25-(OH)2-D3 and 1α,24R,25-(OH)3-D3] and the first 24-aminovitamin D3 derivatives [24S-benzoylamino-25-OH-D3 and 24S-benzoylamino-1α,25-(OH)2-D3] are reported. The stereogenic center at C-24 was generated through ultrasonically induced aqueous conjugate addition of a iodide to a dioxolanone or to a oxazolidinone. The vitamin D triene system was constructed using the Lythgoe approach. The synthetic route, which is both short (6 or 7 steps from starting iodide) and efficient (32–45% overall yield), constitutes a practical method for the preparation of 24-functionalized metabolites and analogues of vitamin D3. The ultrasonically induced conjugate addition in the key step provides a novel example of a highly stereoselective reaction promoted by the zinc–copper couple in aqueous media.
Suárez, R. M.; Pérez Sestelo, J.; Sarandeses, L. A.
Chem. Eur. J. 2003, 9, 4179–4187 (DOI: 10.1002/chem.200304790).
The stereoselectivity of the ultrasonically induced zinc–copper conjugate addition of iodides to chiral α,β-unsaturated carbonyl systems under aqueous conditions was studied. Alkyl iodides add diastereoselectively to a methylenedioxolanone and to a methyleneoxazolidinone to afford the 1,4-addition products in good yields (38–95%) and with high diastereomeric excess (44–90% de). The 1,4-addition to chiral γ,δ-dioxolanyl-α,β-unsaturated esters also proceeds with good yields (51–99%). The diastereoselectivity is dependent on the geometry of the olefin: the Z isomer gives high diastereoselectivity, while the reactions with the E isomer are nonstereoselective. The reaction proceeds with excellent chemoselectivity and allows the use of iodides bearing ester, hydroxy, and amino groups. Since the 1,4-addition products can be readily hydrolyzed, this methodology constitutes a novel entry for the enantioselective synthesis of α-and γ-hydroxy acids and α-amino acids in aqueous media. The results obtained support the radical mechanism proposed by Luche, and represent one of the few examples of a radical stereoselective conjugate addition in aqueous medium.
Pena, M. A.; Pérez Sestelo, J.; Sarandeses, L. A.
Synthesis 2003, 780–784 (DOI: 10.1055/s-2003-38060).
A novel palladium-catalyzed carbonylative cross-coupling reaction that employs triorganoindium compounds is described. The reaction proceeds under a carbon monoxide atmosphere with alkyl-, alkynyl-, and arylindium reagents to give good yields of unsymmetrical ketones with high atom economy. All three organic groups attached to the metal are efficiently transferred in this carbonylative coupling reaction.
Rodríguez, D.; Pérez Sestelo, J.; Sarandeses, L. A.
J. Org. Chem. 2003, 68, 2518–2520 (DOI: 10.1021/jo0265939).
The first nucleophilic allylic substitution reactions of triorganoindium compounds with allylic halides and phosphates are reported. The reactions of trialkyl- and triarylindium reagents with cinnamyl and geranyl halides and phosphates, with the aid of copper catalysis [Cu(OTf)2/P(OEt)3], are described. In general, the reaction proceeds efficiently to give good yields and regioselectively to afford the SN2’ product.
Real, M. M.; Pérez Sestelo, J.; Sarandeses, L. A.
Tetrahedron Lett. 2002, 43, 9111–9114 (DOI: 10.1016/S0040-4039(02)02246-3).
Functionalized benzo[c]phenanthrenes ([4]helicenes) and [5]helicenes were synthesised in five steps from tetrahydronaphthalenone and tetrahydrophenanthrenone compounds using a novel synthetic approach based on a Diels–Alder reaction between inner–outer ring 1,3-bis(trimethylsilyloxy)-1,3-dienes and benzyne or quinones.
Pena, M. A.; Pérez, I.; Pérez Sestelo, J.; Sarandeses, L. A.
Chem. Commun. 2002, 2246–2247 (DOI: 10.1039/b206346h).
Multifold and sequential palladium-catalyzed cross-coupling reactions can be performed between triorganoindium compounds and oligohaloarenes using only a small excess of the organometallic reagent, low catalyst charge loading and short reaction times.
Suárez, R. M.; Pérez Sestelo, J.; Sarandeses, L. A.
Synlett 2002, 1435–1438 (DOI: 10.1055/s-2002-33604).
The diastereoselective ultrasonically induced zinc–copper 1,4-addition of alkyl iodides to chiral α,β-unsaturated systems in aqueous media is reported. The reaction of a methylenedioxolanone with a variety of alkyl iodides takes place in good yields and with high diastereomeric excess. The 1,4-addition to (Z)-and (E)-γ,δ-dioxolanyl-a,b-unsaturated esters also proceeds with good yields: the Z-isomer gives good diastereoselectivities while the reactions with the E-isomer are non-stereoselective.
Cornella, I.; Pérez Sestelo, J.; Mouriño, A.; Sarandeses, L. A.
J. Org. Chem. 2002, 67, 4707–4714 (DOI: 10.1021/jo020022z).
A novel convergent synthetic approach to new analogues of calcitriol modified at the C-18 position is reported. The key step in the synthesis is the 20-hydroxyl-directed photochemical iodination of the 18-methyl group in the presence of (diacetoxyiodo)benzene. Using this methodology, two new analogues of calcitriol were prepared: the first contains a hydroxylated alkyl side chain attached at C-18 with the natural side chain replaced by an isopropylidene group; the second is a conformationally locked analogue due to an extra oxacycle between the C-18 and C-20 positions.
Pérez Sestelo, J.; Cornella, I.; de Uña, O.; Mouriño, A.; Sarandeses, L. A.
Chem. Eur. J. 2002, 8, 2747–2752 (DOI: 10.1002/1521-3765(20020617)8:12<2747::AID-CHEM2747>3.0.CO;2-J).
Vitamin D3 active metabolites 24R,25-(OH)2-D3, 24S,25-(OH)2-D3 , and 1a,24R,25-(OH)3-D3 were synthesized by a convergent and stereoselective approach. In the synthetic route, the stereogenic center at C-24 was generated through ultrasonically induced aqueous conjugate addition of a iodide to Seebach’s dioxolanone, and the vitamin D triene system was constructed using the Lythgoe approach.
The synthesis, which is both short (seven steps from iodide) and efficient (32–40% overall yield), allows the preparation of large quantities of the metabolites and provides a novel example of a highly stereoselective reaction promoted by the zinc-copper couple in aqueous media.
Pérez Sestelo, J.; de Uña, O.; Mouriño, A.; Sarandeses, L. A.
Synlett 2002, 719–722 (DOI: 10.1055/s-2002-25339).
The synthesis of the first 24-aminovitamin D3 derivatives is reported. A stereoselective convergent synthetic approach was employed to prepare 24(S)-benzoylamino-25-hydroxyvitamin D3 and 24(S)-benzoylamino-1a,25-dihydroxyvitamin D3 in six steps from iodide 2 in 34% and 42% overall yields, respectively. The key step in the synthesis is a novel diastereoselective ultrasonically induced conjugate addition, promoted by the zinc–copper couple, of iodide 2 to a methyleneoxazolidinone in aqueous media. The conjugate vitamin D triene system was constructed using the Lythgoe approach.
Pérez, I.; Pérez Sestelo, J.; Sarandeses, L. A.
J. Am. Chem. Soc. 2001, 123, 4155–4160 (DOI: 10.1021/ja004195).
The novel metal-catalyzed cross-coupling reaction of indium organometallics with organic electrophiles is described. Triorganoindium compounds (R3In) containing alkyl, vinyl, aryl, and alkynyl groups are efficiently prepared from the corresponding lithium or magnesium organometallics by reaction with indium trichloride. The cross-coupling reaction of R3In with aryl halides and pseudohalides, vinyl triflates, benzyl bromides, and acid chlorides proceeds under palladium catalysis in excellent yields and with high chemoselectivity. Indium organometallics also react with aryl chlorides as under nickel catalysis. In the cross-coupling reaction the triorganoindium compounds transfer, in a clear example of atom economy, all three of the organic groups attached to the metal, as shown by the necessity of using only 34 mol% of indium. The feasibility of using R3In in reactions with different electrophiles, along with the high yields and chemoselectivities obtained, reveals indium organometallics to be useful alternatives to other organometallics in cross-coupling reactions.
Pérez Sestelo, J.; Real, M. M.; Sarandeses, L. A.
J. Org. Chem. 2001, 66, 1395–1402 (DOI: 10.1021/jo0015319).
A Diels-Alder reaction of novel inner-outer-ring 1,3-silyloxydienes with a variety of dienophiles to afford highly functionalized polycyclic structures is reported. The inner-outer-ring 1,3-silyloxydienes containing five- to seven-membered carbocyclic and heterocyclic rings were prepared in a single reaction vessel from 2-acetylcyclocarbonyls in quantitative yields. The Diels-Alder reaction with 1,4-benzoquinone (BQ), dimethyl acetylenedicarboxylate (DMAD), and methyl vinyl ketone (MVK) proceeded smoothly at room temperature, affording functionalized polycyclic naphthols, phenols, and enones with high regioselectivity and good yields (39-75%). Moreover, these dienes also reacted in a hetero-Diels-Alder reaction with benzaldehyde (BA) and N-benzylideneaniline (NBA) in the presence of catalytic amounts of ZnCl2, affording substituted polycyclic pyranones and pyridinones in good yields (40-93%). Overall, our synthetic strategy provides straightforward access to an interesting set of polycyclic structures useful for natural and nonnatural product synthesis.
-2000
Pérez Sestelo, J.; Mouriño, A.; Sarandeses, L. A.
J. Org. Chem. 2000, 65, 8290–8296 (DOI: 10.1021/jo001084x).
The design and synthesis of vitamin D3 dimers and 1a,25-dihydroxyvitamin D3 (calcitriol) dimers are described. The dimers were designed with a view to doubly binding the vitamin D receptor (VDR) and inducing the receptor homodimerization. In the dimers the units are linked through the C-11 position in ring C by an alkyl side chain of six or 10 carbon atoms, far from the hydroxy groups responsible for the VDR binding. The linker is formed by olefin metathesis of an olefinic side chain at the C-11 position introduced by stereoselective cuprate addition. The synthesis, which is both short and convergent, uses the Wittig-Horner approach to construct the vitamin D triene system and allows the preparation of dimers with a linker of modulated length with the purpose of optimizing the vitamin D3-VDR interaction.
Pérez, I.; Pérez Sestelo, J.; Sarandeses, L. A.
Org. Lett. 1999, 1, 1267–1269 (DOI: 10.1021/ol990939t).
A novel palladium-catalyzed cross-coupling reaction of organoindium compounds with vinyl and aryl triflates or iodides is described. The. reaction proceeds for alkyl-, vinyl-, alkynyl-, and arylindium compounds in excellent yields and high chemoselectivity without any excess of the organometallic. Remarkably, indium organometallics transfer efficiently the three organic groups attached to the metal.
Pérez Sestelo, J.; Mouriño, A.; Sarandeses, L. A.
Org. Lett. 1999, 1, 1005–1007 (DOI: 10.1021/ol990878z).
A dimer comprising two 1a,25-dihydroxyvitamin D3 units linked by an alkyl side chain at C-11 was synthesized with a view to the simultaneous binding of two vitamin D receptor (VDR) molecules and the consequent induction of VDR dimerization. The short, convergent synthesis uses a stereoselective cuprate addition to introduce the linking side chain and a key ruthenium olefin metathesis as the dimerization step.
Pérez Sestelo, J.; Real, M. M.; Mouriño, A.; Sarandeses, L. A.
Tetrahedron Lett. 1999, 40, 985–988 (DOI: 10.1016/S0040-4039(98)02463-0).
In this paper we describe regioselective one-pot preparations of inner-outer-ring 1,3-bis[trimethylsilyloxy]dienes and their Diels-Alder reactions with electron-deficient dienophiles. The dienes were prepared in quantitative yield, and the Diels-Alder reactions proceeded smoothly, regioselectively and with good yields, allowing efficient construction of highly functionalized polycyclic skeletons such as naphthofurans, benzofurans and various carbocycles present in natural and non-natural products.
Pérez, I.; Pérez Sestelo, J.; Maestro, M. A.; Mouriño, A.; Sarandeses, L. A.
J. Org. Chem. 1998, 63, 10074–10076 (DOI: 10.1021/jo981830m).
A novel, efficient nickel-catalyzed 1,4-conjugate addition of triorganoindium compounds to ,-unsaturated systems is described. Best results were obtained using Ni(COD)2. The range of groups susceptible of addition was studied using aryl and primary and tertiary alkyl organoindium compounds prepared from commercially available organolithium compounds by transmetallation with InCl3. Addition to enones, esters and nitriles occurred in yields ranging from 50 to 89%.